| 98. |
Karyotypic Profiling of Induced Pluripotent Stem Cells Derived from a Xeroderma Pigmentosum Group C Patient.
Alsalloum A, Mingaleva N, Gornostal E, Antysheva Z, Sparber P, Skoblov M, Pozhitnova V, Belysheva T, Levashova A, Kuznetsova E, Suvorova Y, Krupinova J, Bogdanov V, Abyzov A, Mityaeva O, Volchkov P
Cells 2025; 14(24):1985
 
|
| 97. |
The Somatic Mosaicism across Human Tissues Network.
Coorens THH, Oh JW, Choi YA, Lim NS, Zhao B, Voshall A, Abyzov A, Antonacci-Fulton L, Aparicio S, Ardlie KG, Bell TJ, Bennett JT, Bernstein BE, Blanchard TG, Boyle AP, Buenrostro JD, Burns KH, Chen F, Chen R, Choudhury S, Doddapaneni HV, Eichler EE, Evrony GD, Faith MA, Fazzio TG, Fulton RS, Garber M, Gehlenborg N, Germer S, Getz G, Gibbs RA, Hernandez RG, Jin F, Korbel JO, Landau DA, Lawson HA, Lennon NJ, Li H, Li Y, Loh PR, Marth G, McConnell MJ, Mills RE, Montgomery SB, Natarajan P, Park PJ, Satija R, Sedlazeck FJ, Shao DD, Shen H, Stergachis AB, Underhill HR, Urban AE, VonDran MW, Walsh CA, Wang T, Wu TP, Zong C, Lee EA, Vaccarino FM
Nature 2025; 643(8070):47-59

|
| 96. |
Specification of human brain regions with orthogonal gradients of WNT and SHH in organoids reveals patterning variations across cell lines.
Scuderi S, Kang TY, Jourdon A, Nelson A, Yang L, Wu F, Anderson GM, Mariani J, Tomasini L, Sarangi V, Abyzov A, Levchenko A, Vaccarino FM
Cell Stem Cell 2025; 32(6):970-989.e11
 
|
| 95. |
Interneuron Loss and Microglia Activation by Transcriptome Analyses in the Basal Ganglia of Tourette Disorder.
Wang Y, Fasching L, Wu F, Suvakov M, Huttner A, Berretta S, Roberts R, Leckman JF, Fernandez TV, Abyzov A, Vaccarino FM
Biol Psychiatry 2025; 98(3):260-270

|
| 94. |
Transgenerational transmission of post-zygotic mutations suggests symmetric contribution of first two blastomeres to human germline.
Jang Y, Tomasini L, Bae T, Szekely A, Vaccarino FM, Abyzov A
Nat Commun 2024; 15(1):9117
|
| 93. |
Somatic mosaicism in schizophrenia brains reveals prenatal mutational processes.
Maury EA, Jones A, Seplyarskiy V, Nguyen TTL, Rosenbluh C, Bae T, Wang Y, Abyzov A, Khoshkhoo S, Chahine Y, Zhao S, Venkatesh S, Root E, Voloudakis G, Roussos P, Park PJ, Akbarian S, Brennand K, Reilly S, Lee EA, Sunyaev SR, Walsh CA, Chess A
Science 2024; 386(6718):217-224

|
| 92. |
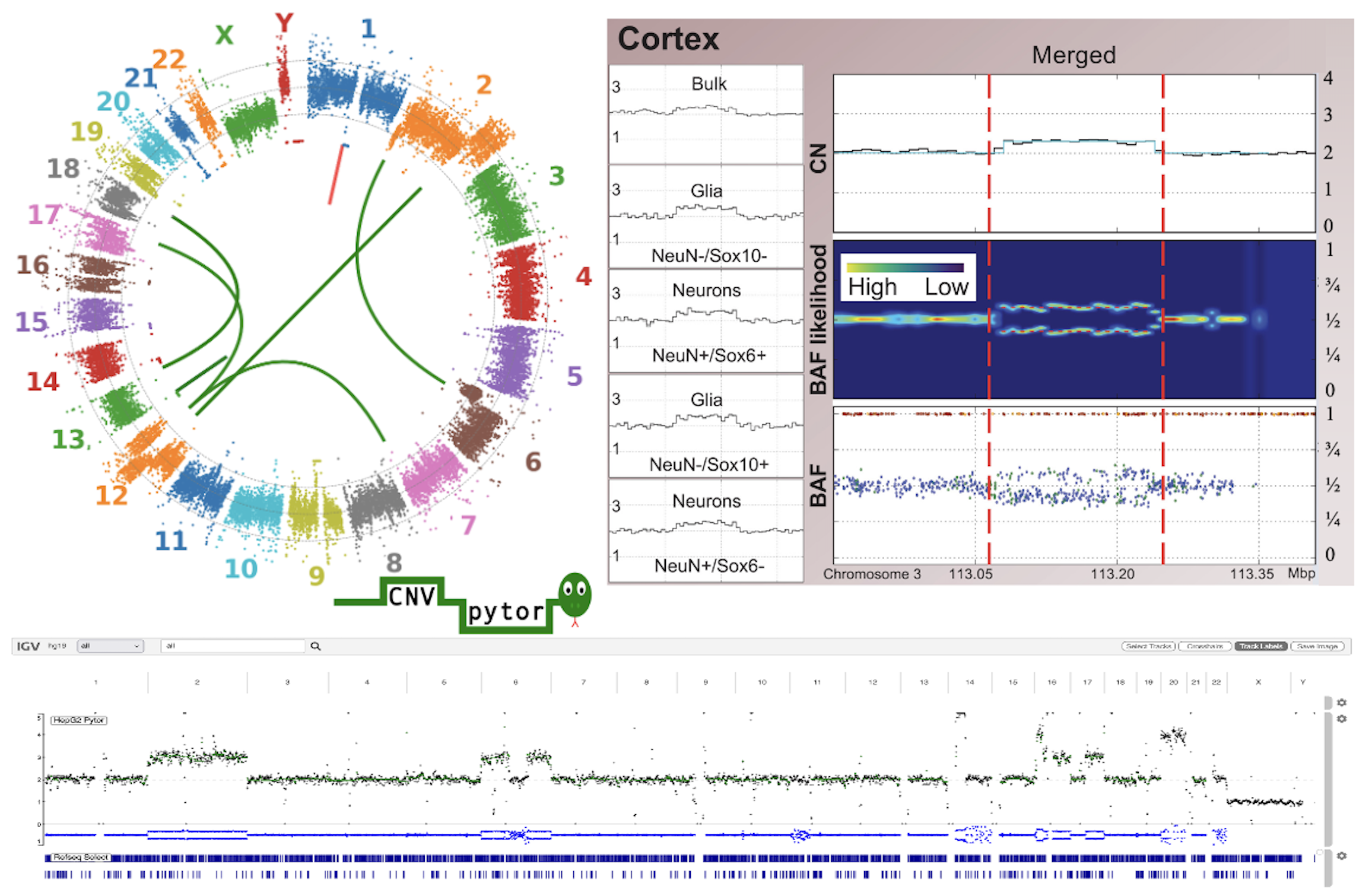
Genome-wide analysis and visualization of copy number with CNVpytor in igv.js.
Panda A, Suvakov M, Thorvaldsdottir H, Mesirov JP, Robinson JT, Abyzov A
Bioinformatics 2024; 40(8):btae453

|
| 91. |
Resolving the 22q11.2 deletion using CTLR-Seq reveals chromosomal rearrangement mechanisms and individual variance in breakpoints.
Zhou B, Purmann C, Guo H, Shin G, Huang Y, Pattni R, Meng Q, Greer SU, Roychowdhury T, Wood RN, Ho M, Dohna HZ, Abyzov A, Hallmayer JF, Wong WH, Ji HP, Urban AE
Proc Natl Acad Sci U S A 2024; 121(31):e2322834121

|
| 90. |
Characterization of enhancer activity in early human neurodevelopment using Massively Parallel Reporter Assay (MPRA) and forebrain organoids.
Capauto D, Wang Y, Wu F, Norton S, Mariani J, Inoue F, Crawford GE, Ahituv N, Abyzov A, Vaccarino FM
Sci Rep 2024; 14(1):3936
|
| 89. |
Genomic data resources of the Brain Somatic Mosaicism Network for neuropsychiatric diseases.
Garrison MA, Jang Y, Bae T, Cherskov A, Emery SB, Fasching L, Jones A, Moldovan JB, Molitor C, Pochareddy S, Peters MA, Shin JH, Wang Y, Yang X, Akbarian S, Chess A, Gage FH, Gleeson JG, Kidd JM, McConnell M, Mills RE, Moran JV, Park PJ, Sestan N, Urban AE, Vaccarino FM, Walsh CA, Weinberger DR, Wheelan SJ, Abyzov A
Sci Data 2023; 10(1):813
|
| 88. |
Modeling idiopathic autism in forebrain organoids reveals an imbalance of excitatory cortical neuron subtypes during early neurogenesis.
Jourdon A, Wu F, Mariani J, Capauto D, Norton S, Tomasini L, Amiri A, Suvakov M, Schreiner JD, Jang Y, Panda A, Nguyen CK, Cummings EM, Han G, Powell K, Szekely A, McPartland JC, Pelphrey K, Chawarska K, Ventola P, Abyzov A, Vaccarino FM
Nat Neurosci 2023; 26(9):1505-1515
|
| 87. |
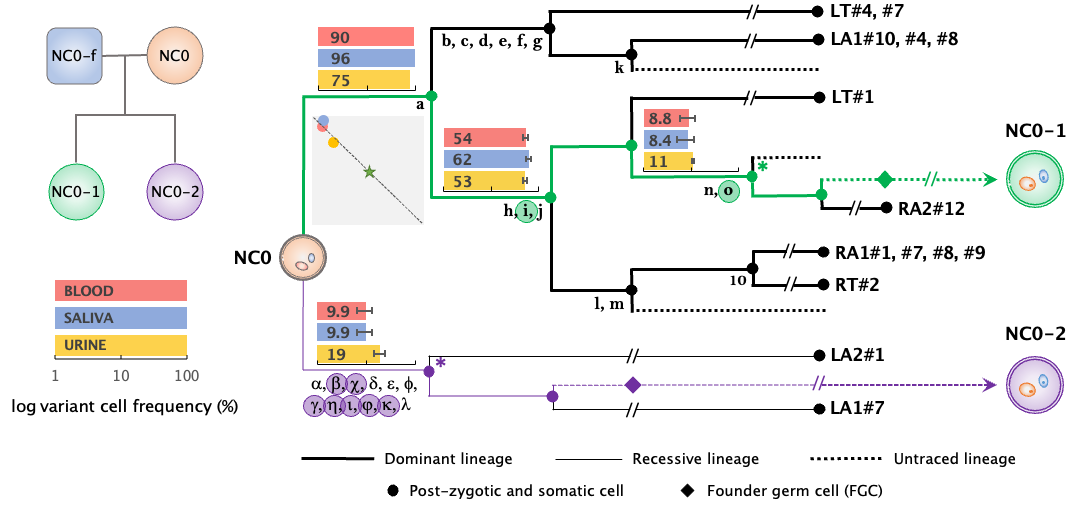
Efficient reconstruction of cell lineage trees for cell ancestry and cancer.
Jang Y, Fasching L, Bae T, Tomasini L, Schreiner J, Szekely A, Fernandez TV, Leckman JF, Vaccarino FM, Abyzov A
Nucleic Acids Res 2023; 51(10):e57
|
| 86. |
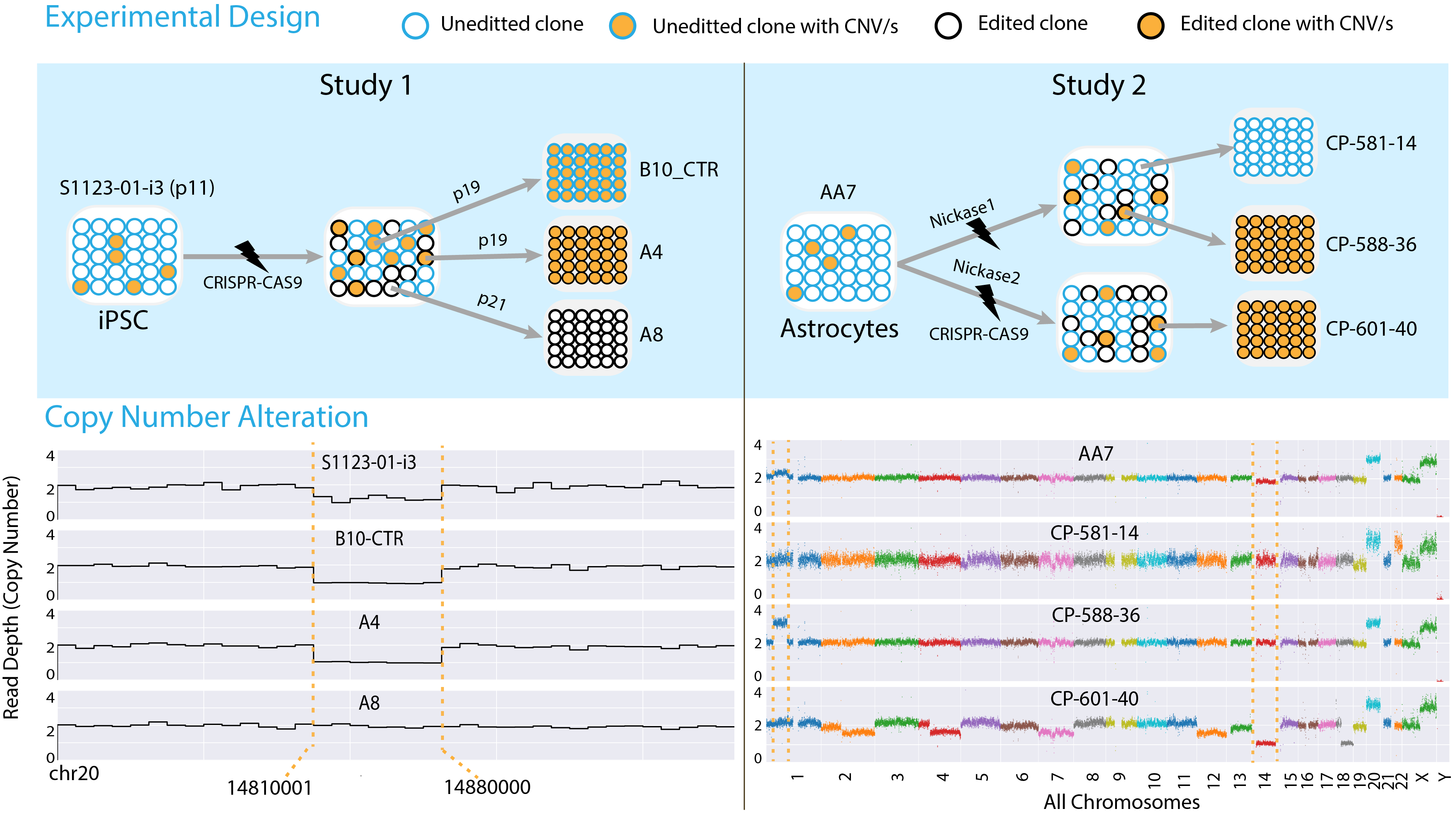
Clonally Selected Lines After CRISPR-Cas Editing Are Not Isogenic.
Panda A, Suvakov M, Mariani J, Drucker KL, Park Y, Jang Y, Kollmeyer TM, Sarkar G, Bae T, Kim JJ, Yoon WH, Jenkins RB, Vaccarino FM, Abyzov A
CRISPR J 2023; 6(2):176-182

|
| 85. |
Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development.
Chung C, Yang X, Bae T, Vong KI, Mittal S, Donkels C, Westley Phillips H, Li Z, Marsh APL, Breuss MW, Ball LL, Garcia CAB, George RD, Gu J, Xu M, Barrows C, James KN, Stanley V, Nidhiry AS, Khoury S, Howe G, Riley E, Xu X, Copeland B, Wang Y, Kim SH, Kang HC, Schulze-Bonhage A, Haas CA, Urbach H, Prinz M, Limbrick DD Jr, Gurnett CA, Smyth MD, Sattar S, Nespeca M, Gonda DD, Imai K, Takahashi Y, Chen HH, Tsai JW, Conti V, Guerrini R, Devinsky O, Silva WA Jr, Machado HR, Mathern GW, Abyzov A, Baldassari S, Baulac S, Gleeson JG
Nat Genet 2023; 55(2):209-220
|
| 84. |
Control-independent mosaic single nucleotide variant detection with DeepMosaic.
Yang X, Xu X, Breuss MW, Antaki D, Ball LL, Chung C, Shen J, Li C, George RD, Wang Y, Bae T, Cheng Y, Abyzov A, Wei L, Alexandrov LB, Sebat JL, Gleeson JG
Nat Biotechnol 2023; 41(6):870-877
|
| 83. |
OpBerg: Discovering Causal Sentences Using Optimal Alignments.
Wood J, Matiasz N, Silva A, Hsu W, Abyzov A, Wang W, Wrembel R, Gamper J, Kotsis G, Tjoa A, Khalil I
Cham: Springer International Publishing 2022;
|
| 82. |
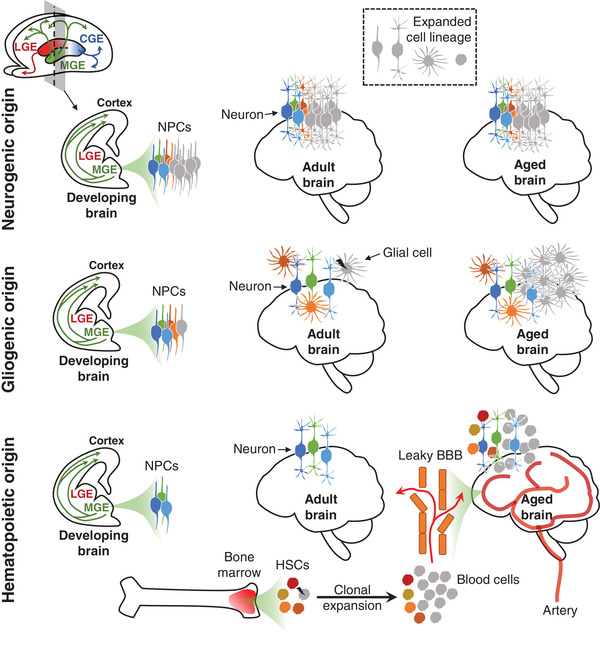
Somatic genomic mosaicism in the brain during aging: Scratching the surface.
Bae T, Wang Y, Vaccarino FM, Abyzov A
Clin Transl Med 2022; 12(12):e1138
|
| 81. |
A noncoding single-nucleotide polymorphism at 8q24 drives IDH1-mutant glioma formation.
Yanchus C, Drucker KL, Kollmeyer TM, Tsai R, Winick-Ng W, Liang M, Malik A, Pawling J, De Lorenzo SB, Ali A, Decker PA, Kosel ML, Panda A, Al-Zahrani KN, Jiang L, Browning JWL, Lowden C, Geuenich M, Hernandez JJ, Gosio JT, Ahmed M, Loganathan SK, Berman J, Trcka D, Michealraj KA, Fortin J, Carson B, Hollingsworth EW, Jacinto S, Mazrooei P, Zhou L, Elia A, Lupien M, He HH, Murphy DJ, Wang L, Abyzov A, Dennis JW, Maass PG, Campbell K, Wilson MD, Lachance DH, Wrensch M, Wiencke J, Mak T, Pennacchio LA, Dickel DE, Visel A, Wrana J, Taylor MD, Zadeh G, Dirks P, Eckel-Passow JE, Attisano L, Pombo A, Ida CM, Kvon EZ, Jenkins RB, Schramek D
Science 2022; 378(6615):68-78
|
| 80. |
Analysis of somatic mutations in 131 human brains reveals aging-associated hypermutability.
Bae T, Fasching L, Wang Y, Shin JH, Suvakov M, Jang Y, Norton S, Dias C, Mariani J, Jourdon A, Wu F, Panda A, Pattni R, Chahine Y, Yeh R, Roberts RC, Huttner A, Kleinman JE, Hyde TM, Straub RE, Walsh CA, Urban AE, Leckman JF, Weinberger DR, Vaccarino FM, Abyzov A
Science 2022; 377(6605):511-517
|
| 79. |
Postmortem Human Dura Mater Cells Exhibit Phenotypic, Transcriptomic and Genetic Abnormalities that Impact their Use for Disease Modeling.
Argouarch AR, Schultz N, Yang AC, Jang Y, Garcia K, Cosme CG, Corrales CI, Nana AL, Karydas AM, Spina S, Grinberg LT, Miller B, Wyss-Coray T, Abyzov A, Goodarzi H, Seeley WW, Kao AW
Stem Cell Rev Rep 2022; 18(8):3050-3065

|
| 78. |
All2: A tool for selecting mosaic mutations from comprehensive multi-cell comparisons.
Sarangi V, Jang Y, Suvakov M, Bae T, Fasching L, Sekar S, Tomasini L, Mariani J, Vaccarino FM, Abyzov A
PLoS Comput Biol 2022; 18(4):e1009487

|
| 77. |
CNVpytor: a tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing.
Suvakov M, Panda A, Diesh C, Holmes I, Abyzov A
Gigascience 2021; 10(11):giab074
|
| 76. |
LongAGE: defining breakpoints of genomic structural variants through optimal and memory efficient alignments of long reads.
Tran Q, Abyzov A
Bioinformatics 2021; 37(7):1015-1017
|
| 75. |
Comprehensive identification of somatic nucleotide variants in human brain tissue.
Wang Y, Bae T, Thorpe J, Sherman MA, Jones AG, Cho S, Daily K, Dou Y, Ganz J, Galor A, Lobon I, Pattni R, Rosenbluh C, Tomasi S, Tomasini L, Yang X, Zhou B, Akbarian S, Ball LL, Bizzotto S, Emery SB, Doan R, Fasching L, Jang Y, Juan D, Lizano E, Luquette LJ, Moldovan JB, Narurkar R, Oetjens MT, Rodin RE, Sekar S, Shin JH, Soriano E, Straub RE, Zhou W, Chess A, Gleeson JG, Marquès-Bonet T, Park PJ, Peters MA, Pevsner J, Walsh CA, Weinberger DR, Vaccarino FM, Moran JV, Urban AE, Kidd JM, Mills RE, Abyzov A
Genome Biol 2021; 22(1):92

|
| 74. |
Early developmental asymmetries in cell lineage trees in living individuals.
Fasching L, Jang Y, Tomasi S, Schreiner J, Tomasini L, Brady MV, Bae T, Sarangi V, Vasmatzis N, Wang Y, Szekely A, Fernandez TV, Leckman JF, Abyzov A, Vaccarino FM
Science 2021; 371(6535):1245-1248
|
| 73. |
Landmarks of human embryonic development inscribed in somatic mutations.
Bizzotto S, Dou Y, Ganz J, Doan RN, Kwon M, Bohrson CL, Kim SN, Bae T, Abyzov A, Park PJ, Walsh CA
Science 2021; 371(6535):1249-1253
|
| 72. |
Machine learning reveals bilateral distribution of somatic L1 insertions in human neurons and glia.
Zhu X, Zhou B, Pattni R, Gleason K, Tan C, Kalinowski A, Sloan S, Fiston-Lavier AS, Mariani J, Petrov D, Barres BA, Duncan L, Abyzov A, Vogel H, Moran JV, Vaccarino FM, Tamminga CA, Levinson DF, Urban AE
Nat Neurosci 2021; 24(2):186-196
|
| 71. |
PsychENCODE and beyond: transcriptomics and epigenomics of brain development and organoids.
Jourdon A, Scuderi S, Capauto D, Abyzov A, Vaccarino FM
Neuropsychopharmacology 2021; 46(1):70-85
|
| 70. |
Analysis of Cell and Nucleus Genome by Next-Generation Sequencing [Internet]
Oh J, Abyzov A
Cham: Springer International Publishing; 2020; 35-65p
|
| 69. |
Adult diffuse glioma GWAS by molecular subtype identifies variants in D2HGDH and FAM20C.
Eckel-Passow JE, Drucker KL, Kollmeyer TM, Kosel ML, Decker PA, Molinaro AM, Rice T, Praska CE, Clark L, Caron A, Abyzov A, Batzler A, Song JS, Pekmezci M, Hansen HM, McCoy LS, Bracci PM, Wiemels J, Wiencke JK, Francis S, Burns TC, Giannini C, Lachance DH, Wrensch M, Jenkins RB
Neuro Oncol 2020; 22(11):1602-1613
|
| 68. |
SCELLECTOR: ranking amplification bias in single cells using shallow sequencing.
Sarangi V, Jourdon A, Bae T, Panda A, Vaccarino F, Abyzov A
BMC Bioinformatics 2020; 21(1):521
|
| 67. |
Complex mosaic structural variations in human fetal brains.
Sekar S, Tomasini L, Proukakis C, Bae T, Manlove L, Jang Y, Scuderi S, Zhou B, Kalyva M, Amiri A, Mariani J, Sedlazeck FJ, Urban AE, Vaccarino FM, Abyzov A
Genome Res 2020; 30(12):1695-1704

|
| 66. |
The role of somatic mosaicism in brain disease.
Jourdon A, Fasching L, Scuderi S, Abyzov A, Vaccarino FM
Curr Opin Genet Dev 2020; [Epub ahead of print]
|
| 65. |
Cell Lineage Tracing and Cellular Diversity in Humans.
Abyzov A, Vaccarino FM
Annu Rev Genomics Hum Genet 2020; [Epub ahead of print]

|
| 64. |
Neurological safety of oxaliplatin in patients with uncommon variants in Charcot-Marie-tooth disease genes.
Le-Rademacher JG, Lopez CL, Kanwar R, Major-Elechi B, Abyzov A, Banck MS, Therneau TM, Sloan JA, Loprinzi CL, Beutler AS
J Neurol Sci 2020; [Epub ahead of print]
|
| 63. |
Combining copy number, methylation markers, and mutations as a panel for endometrial cancer detection via intravaginal tampon collection.
Sangtani A, Wang C, Weaver A, Hoppman NL, Kerr SE, Abyzov A, Shridhar V, Staub J, Kocher JA, Voss JS, Podratz KC, Wentzensen N, Kisiel JB, Sherman ME, Bakkum-Gamez JN
Gynecol Oncol 2020; 156(2):387-392
|
| 62. |
Approaches and Methods for Variant Analysis in the Genome of a Single Cell [Internet]
Abyzov A, Vaccarino F, Urban A, Sarangi V
Cham: Springer International Publishing; 2019; 203-228p
|
| 61. |
Haplotype-resolved and integrated genome analysis of the cancer cell line HepG2.
Zhou B, Ho SS, Greer SU, Spies N, Bell JM, Zhang X, Zhu X, Arthur JG, Byeon S, Pattni R, Saha I, Huang Y, Song G, Perrin D, Wong WH, Ji HP, Abyzov A, Urban AE
Nucleic Acids Res 2019; 47(8):3846-3861
|
| 60. |
Chromatin organization modulates the origin of heritable structural variations in human genome.
Roychowdhury T, Abyzov A
Nucleic Acids Res 2019; 47(6):2766-2777
|
| 59. |
Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562.
Zhou B, Ho SS, Greer SU, Zhu X, Bell JM, Arthur JG, Spies N, Zhang X, Byeon S, Pattni R, Ben-Efraim N, Haney MS, Haraksingh RR, Song G, Ji HP, Perrin D, Wong WH, Abyzov A, Urban AE
Genome Res 2019; 29(3):472-484
|
| 58. |
Molecular signatures of multiple myeloma progression through single cell RNA-Seq.
Jang JS, Li Y, Mitra AK, Bi L, Abyzov A, van Wijnen AJ, Baughn LB, Van Ness B, Rajkumar V, Kumar S, Jen J
Blood Cancer J 2019; 9(1):2
|
| 57. |
Detection and Quantification of Mosaic Genomic DNA Variation in Primary Somatic Tissues Using ddPCR: Analysis of Mosaic Transposable-Element Insertions, Copy-Number Variants, and Single-Nucleotide Variants.
Zhou B, Haney MS, Zhu X, Pattni R, Abyzov A, Urban AE
Methods Mol Biol 2018; [Epub ahead of print]

|
| 56. |
Revealing the brain's molecular architecture.
Science 2018; 362(6420):1262-1263
|
| 55. |
Transcriptome and epigenome landscape of human cortical development modeled in organoids.
Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, Pochareddy S, Shin Y, Safi A, Song L, Zhu Y, Sousa AMM, Gerstein M, Crawford GE, Sestan N, Abyzov A, Vaccarino FM
Science 2018; 362(6420):eaat6720
|
| 54. |
Molecular characterization of colorectal adenomas with and without malignancy reveals distinguishing genome, transcriptome and methylome alterations.
Druliner BR, Wang P, Bae T, Baheti S, Slettedahl S, Mahoney D, Vasmatzis N, Xu H, Kim M, Bockol M, O'Brien D, Grill D, Warner N, Munoz-Gomez M, Kossick K, Johnson R, Mouchli M, Felmlee-Devine D, Washechek-Aletto J, Smyrk T, Oberg A, Wang J, Chia N, Abyzov A, Ahlquist D, Boardman LA
Sci Rep 2018; 8(1):3161
|
| 53. |
Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis.
Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, Riley-Gillis B, Sestan N, Urban AE, Abyzov A, Vaccarino FM
Science 2018; 359(6375):550-555
|
| 52. |
Genomic Mosaicism in Neurons and Other Cell Types
Abyzov A, Urban AE, Vaccarino FM
Principles and Approaches for Discovery and Validation of Somatic Mosaicism in the Human Brain., Springer New York: Springer Nature; 2017; Chapter 1.; 3-24p
|
| 51. |
Inferring modes of evolution from colorectal cancer with residual polyp of origin.
Kim M, Druliner BR, Vasmatzis N, Bae T, Chia N, Abyzov A, Boardman LA
Oncotarget 2017; 9(6):6780-6792

|
| 50. |
Patient-reported (EORTC QLQ-CIPN20) versus physician-reported (CTCAE) quantification of oxaliplatin- and paclitaxel/carboplatin-induced peripheral neuropathy in NCCTG/Alliance clinical trials.
Le-Rademacher J, Kanwar R, Seisler D, Pachman DR, Qin R, Abyzov A, Ruddy KJ, Banck MS, Lavoie Smith EM, Dorsey SG, Aaronson NK, Sloan J, Loprinzi CL, Beutler AS
Support Care Cancer 2017; 25(11):3537-3544
|
| 49. |
Landscape and variation of novel retroduplications in 26 human populations.
Zhang Y, Li S, Abyzov A, Gerstein MB
PLoS Comput Biol 2017; 13(6):e1005567
|
| 48. |
Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network.
McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, Ganz J, Jaffe AE, Kwan KY, Kwon M, Lodato MA, Mills RE, Paquola ACM, Rodin RE, Rosenbluh C, Sestan N, Sherman MA, Shin JH, Song S, Straub RE, Thorpe J, Weinberger DR, Urban AE, Zhou B, Gage FH, Lehner T, Senthil G, Walsh CA, Chess A, Courchesne E, Gleeson JG, Kidd JM, Park PJ, Pevsner J, Vaccarino FM
Science 2017; 356(6336):eaal1641
|
| 47. |
Comprehensive performance comparison of high-resolution array platforms for genome-wide Copy Number Variation (CNV) analysis in humans.
Haraksingh RR, Abyzov A, Urban AE
BMC Genomics 2017; 18(1):321
|
| 46. |
Human induced pluripotent stem cells for modelling neurodevelopmental disorders.
Ardhanareeswaran K, Mariani J, Coppola G, Abyzov A, Vaccarino FM
Nat Rev Neurol 2017; 13(5):265-278
|
| 45. |
One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin.
Abyzov A, Tomasini L, Zhou B, Vasmatzis N, Coppola G, Amenduni M, Pattni R, Wilson M, Gerstein M, Weissman S, Urban AE, Vaccarino FM
Genome Res 2017; 27(4):512-523
|
| 44. |
Colorectal Cancer with Residual Polyp of Origin: A Model of Malignant Transformation.
Druliner BR, Rashtak S, Ruan X, Bae T, Vasmatzis N, O'Brien D, Johnson R, Felmlee-Devine D, Washechek-Aletto J, Basu N, Liu H, Smyrk T, Abyzov A, Boardman LA
Transl Oncol 2016; 9(4):280-6

|
| 43. |
Elevated variant density around SV breakpoints in germline lineage lends support to error-prone replication hypothesis.
Dhokarh D, Abyzov A
Genome Res 2016; 26(7):874-81
|
| 42. |
A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals.
Chen J, Rozowsky J, Galeev TR, Harmanci A, Kitchen R, Bedford J, Abyzov A, Kong Y, Regan L, Gerstein M
Nat Commun 2016; [Epub ahead of print]
|
| 41. |
Understanding genome structural variations.
Abyzov A, Li S, Gerstein MB
Oncotarget 2016; 7(7):7370-1
|
| 40. |
Testing of candidate single nucleotide variants associated with paclitaxel neuropathy in the trial NCCTG N08C1 (Alliance).
Boora GK, Kanwar R, Kulkarni AA, Abyzov A, Sloan J, Ruddy KJ, Banck MS, Loprinzi CL, Beutler AS
Cancer Med 2016; 5(4):631-9

|
| 39. |
Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors.
Mitra AK, Mukherjee UK, Harding T, Jang JS, Stessman H, Li Y, Abyzov A, Jen J, Kumar S, Rajkumar V, Van Ness B
Leukemia 2016; 30(5):1094-102
|
| 38. |
The PsychENCODE project.
Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, Geschwind DH, Mill J, Nairn AC, Abyzov A, Pochareddy S, Prabhakar S, Weissman S, Sullivan PF, State MW, Weng Z, Peters MA, White KP, Gerstein MB, Amiri A, Armoskus C, Ashley-Koch AE, Bae T, Beckel-Mitchener A, Berman BP, Coetzee GA, Coppola G, Francoeur N, Fromer M, Gao R, Grennan K, Herstein J, Kavanagh DH, Ivanov NA, Jiang Y, Kitchen RR, Kozlenkov A, Kundakovic M, Li M, Li Z, Liu S, Mangravite LM, Mattei E, Markenscoff-Papadimitriou E, Navarro FC, North N, Omberg L, Panchision D, Parikshak N, Poschmann J, Price AJ, Purcaro M, Reddy TE, Roussos P, Schreiner S, Scuderi S, Sebra R, Shibata M, Shieh AW, Skarica M, Sun W, Swarup V, Thomas A, Tsuji J, van Bakel H, Wang D, Wang Y, Wang K, Werling DM, Willsey AJ, Witt H, Won H, Wong CC, Wray GA, Wu EY, Xu X, Yao L, Senthil G, Lehner T, Sklar P, Sestan N
Nat Neurosci 2015; 18(12):1707-12
|
| 37. |
An integrated map of structural variation in 2,504 human genomes.
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, Konkel MK, Malhotra A, Stütz AM, Shi X, Casale FP, Chen J, Hormozdiari F, Dayama G, Chen K, Malig M, Chaisson MJP, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HYK, Mu XJ, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong Z, Clarke L, Dal E, Ding L, Emery S, Fan X, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer EW, McCarthy S, Flicek P, Gibbs RA, Marth G, Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A, Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA, Wang M, Yu F, Zhang C, Zhang J, Zheng-Bradley X, Zhou W, Zichner T, Sebat J, Batzer MA, McCarroll SA, Mills RE, Gerstein MB, Bashir A, Stegle O, Devine SE, Lee C, Eichler EE, Korbel JO
Nature 2015; 526(7571):75-81
|
| 36. |
A global reference for human genetic variation.
Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR
Nature 2015; 526(7571):68-74
|
| 35. |
FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M, Gerstein M, Grigorenko EL, Chawarska K, Pelphrey KA, Howe JR, Vaccarino FM
Cell 2015; 162(2):375-390

|
| 34. |
Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms.
Abyzov A, Li S, Kim DR, Mohiyuddin M, Stütz AM, Parrish NF, Mu XJ, Clark W, Chen K, Hurles M, Korbel JO, Lam HY, Lee C, Gerstein MB
Nat Commun 2015; [Epub ahead of print]
|
| 33. |
MetaSV: an accurate and integrative structural-variant caller for next generation sequencing.
Mohiyuddin M, Mu JC, Li J, Bani Asadi N, Gerstein MB, Abyzov A, Wong WH, Lam HY
Bioinformatics 2015; 31(16):2741-4
|
| 32. |
VarSim: a high-fidelity simulation and validation framework for high-throughput genome sequencing with cancer applications.
Mu JC, Mohiyuddin M, Li J, Bani Asadi N, Gerstein MB, Abyzov A, Wong WH, Lam HY
Bioinformatics 2015; 31(9):1469-71
|
| 31. |
Integrative annotation of variants from 1092 humans: application to cancer genomics.
Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, Das J, Abyzov A, Balasubramanian S, Beal K, Chakravarty D, Challis D, Chen Y, Clarke D, Clarke L, Cunningham F, Evani US, Flicek P, Fragoza R, Garrison E, Gibbs R, Gümüş ZH, Herrero J, Kitabayashi N, Kong Y, Lage K, Liluashvili V, Lipkin SM, MacArthur DG, Marth G, Muzny D, Pers TH, Ritchie GRS, Rosenfeld JA, Sisu C, Wei X, Wilson M, Xue Y, Yu F, Dermitzakis ET, Yu H, Rubin MA, Tyler-Smith C, Gerstein M
Science 2013; 342(6154):1235587
|
| 30. |
Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division.
Abyzov A, Iskow R, Gokcumen O, Radke DW, Balasubramanian S, Pei B, Habegger L, Lee C, Gerstein M
Genome Res 2013; 23(12):2042-52
|
| 29. |
Child development and structural variation in the human genome.
Zhang Y, Haraksingh R, Grubert F, Abyzov A, Gerstein M, Weissman S, Urban AE
Child Dev 2013; 84(1):34-48
|
| 28. |
Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells.
Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, Ferrandino AF, Rosenberg Belmaker LA, Szekely A, Wilson M, Kocabas A, Calixto NE, Grigorenko EL, Huttner A, Chawarska K, Weissman S, Urban AE, Gerstein M, Vaccarino FM
Nature 2012; 492(7429):438-42
|
| 27. |
An integrated map of genetic variation from 1,092 human genomes.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA
Nature 2012; 491(7422):56-65
|
| 26. |
Architecture of the human regulatory network derived from ENCODE data.
Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, Addleman N, Bhardwaj N, Boyle AP, Cayting P, Charos A, Chen DZ, Cheng Y, Clarke D, Eastman C, Euskirchen G, Frietze S, Fu Y, Gertz J, Grubert F, Harmanci A, Jain P, Kasowski M, Lacroute P, Leng JJ, Lian J, Monahan H, O'Geen H, Ouyang Z, Partridge EC, Patacsil D, Pauli F, Raha D, Ramirez L, Reddy TE, Reed B, Shi M, Slifer T, Wang J, Wu L, Yang X, Yip KY, Zilberman-Schapira G, Batzoglou S, Sidow A, Farnham PJ, Myers RM, Weissman SM, Snyder M
Nature 2012; 489(7414):91-100
|
| 25. |
An integrated encyclopedia of DNA elements in the human genome.
Nature 2012; 489(7414):57-74
|
| 24. |
Regulatory element copy number differences shape primate expression profiles.
Iskow RC, Gokcumen O, Abyzov A, Malukiewicz J, Zhu Q, Sukumar AT, Pai AA, Mills RE, Habegger L, Cusanovich DA, Rubel MA, Perry GH, Gerstein M, Stone AC, Gilad Y, Lee C
Proc Natl Acad Sci U S A 2012; 109(31):12656-61
|
| 23. |
Genome-wide mapping of copy number variation in humans: comparative analysis of high resolution array platforms.
Haraksingh RR, Abyzov A, Gerstein M, Urban AE, Snyder M
PLoS One 2011; 6(11):e27859

|
| 22. |
Integration of protein motions with molecular networks reveals different mechanisms for permanent and transient interactions.
Bhardwaj N, Abyzov A, Clarke D, Shou C, Gerstein MB
Protein Sci 2011; 20(10):1745-54
|
| 21. |
AlleleSeq: analysis of allele-specific expression and binding in a network framework.
Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, Bhardwaj N, Rubin M, Snyder M, Gerstein M
Mol Syst Biol 2011; [Epub ahead of print]

|
| 20. |
Identification of genomic indels and structural variations using split reads.
Zhang ZD, Du J, Lam H, Abyzov A, Urban AE, Snyder M, Gerstein M
BMC Genomics 2011; [Epub ahead of print]
|
| 19. |
CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing.
Abyzov A, Urban AE, Snyder M, Gerstein M
Genome Res 2011; 21(6):974-84
|
| 18. |
Mapping copy number variation by population-scale genome sequencing.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stütz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Ye K, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO
Nature 2011; 470(7332):59-65
|
| 17. |
AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision.
Abyzov A, Gerstein M
Bioinformatics 2011; 27(5):595-603
|
| 16. |
Annual Research Review: The promise of stem cell research for neuropsychiatric disorders.
Vaccarino FM, Urban AE, Stevens HE, Szekely A, Abyzov A, Grigorenko EL, Gerstein M, Weissman S
J Child Psychol Psychiatry 2011; 52(4):504-16

|